Research Interests
Our general goal
The main interest of the laboratory is to understand molecular and cellular mechanisms underlying bacterial virulence. In particular, we study processes by which intracellular bacterial pathogens alter the normal functioning of eukaryotic host cells.
Our favourite virulence mechanism
We focus in a virulence mechanism consisting in the one-step injection of bacterial effector proteins into host cells through specialized protein secretion systems. At least the type III, type IV, and type VI secretion systems, found in several Gram-negative bacteria, can function as such protein injection nanodevices. The bacteria we use as experimental models have type III secretion systems (T3SSs; Figure 1) and type IV secretion systems (T4SSs) that are essential for their virulence. Type III and type IV secretion effector proteins have been show to act on a vast array of eukaryotic cell functions, such as cytoskeleton dynamics, cell signalling, vesicular transport, apoptosis, or cell cycle. These effectors are eukaryotic-like proteins because, in activity, function, or structure, they often resemble proteins found in higher organisms.
Our main research interest is to study the function of effector proteins from particular bacterial pathogens (Chlamydia and Legionella).
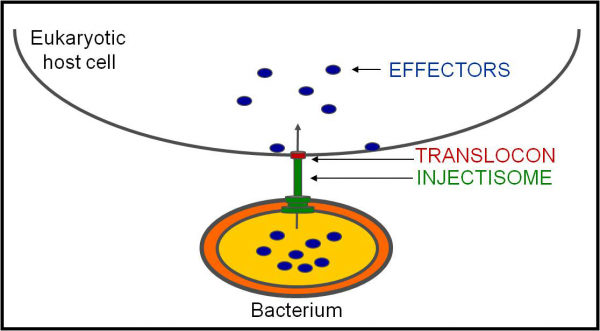
Figure 1. Type III Secretion. Simplified representation of the protein transport mechanism of type III secretion, by which bacteria using a sophisticated apparatus - known as an injectisome, which in many cases is seen in electron microcraphs as a needle-like structure protruding from the bacterial surface - injects effectors into eukaryotic host cells through a translocon pore that is thought to physically link the injectisome needle to a host cell lipid membrane. This enables the delivery of the effector proteins into host cells in a single step. The length of the injectisome needle is tighly controlled by a protein functioning as a molecular ruler. This tight control is needed to overcome the length of other structures at the bacterial surface (e.g. adhesins or LPS) and to enable host cell sensing by the needle tip. Recent years witnessed huge progresses in our knowledge of the structure of the bacterial injectisome.
Our bugs
We focus on intracellular bacterial pathogens that multiply within host cells in unique membrane-bound organelles (pathogen-containing vacuoles), using Chlamydia trachomatis and Legionella pneumophila as experimental models (Figure 2).
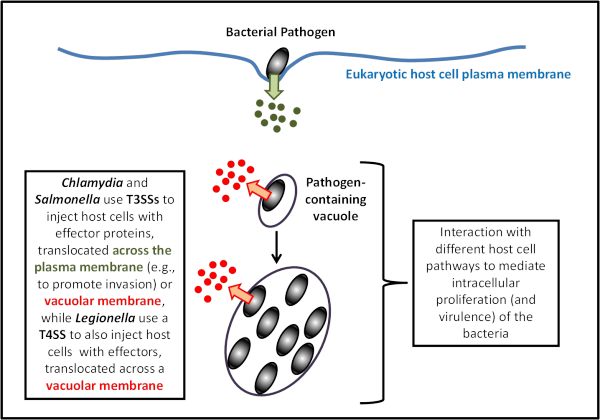
Figure 2. Intravacuolar bacterial pathogens. C. trachomatis (and Salmonella) use T3SSs to invade and replicate within host cells, while L. pneumophila use a T4SS to replicate within host cells.
Chlamydia trachomatis
C. trachomatis is part of a large group of highly related Gram-negative bacteria that are characterized by their obligate growth within eukaryotic cells and which includes the etiological agents of many important human and animal diseases. C. trachomatis causes ocular and genital infections in humans. The ocular serovars infect the conjuctival epithelium and can give rise to trachoma, the leading cause of preventable blindness in developing countries. The genital serovars are the most frequent bacterial cause of sexually transmitted disease worldwide. The genital infections are often asymptomatic and if left untreated they can cause inflammatory diseases which, in turn, can lead to infertility and ectopic pregnancies in women.
Chlamydia are characterised by a unique developmental/infectious cycle that includes two morphological distinct forms: the infectious but metabolically inert elementary bodies (EBs), and the non-infectious but metabolically active reticulate bodies (RBs) (Figure 3).
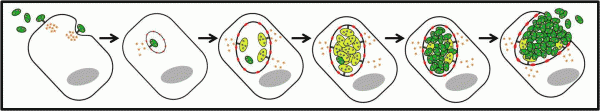
Figure 3. Chlamydia developmental/infectious cycle (image from Sara Pais). Chlamydial elementary bodies (EBs; green) are packed with T3SS effectors and have an assembled injectisome. Upon cell attachment, the effectors (e.g., TARP, CT694/TmeA, and TepP) are delivered into host cells, which is thought to mediate the invasion process. Upon internalization, EBs reside in a membrane bound vacuole and rapidly differentiate into reticulate bodies (RBs; yellow). The initially plasma membrane-derived vacuole is modified by Chlamydia through the T3S of Inclusion membrane (Inc) proteins, which decorate the inclusion membrane throughout the cycle. The RBs replicate by binary fission up to a point where bacterial replication becomes asynchronous and yields both RBs and EBs. Finally, when the inclusion becomes filled with EBs, lysis or extrusion of the host cell occurs and the EBs can start another round of infection.
While Chlamydia has remained genetically intractable for many years, is is now possible to transform, ectopically express proteins, and perform random or site-directed genetic knock-outs in C. trachomatis using group II introns or FRAEM.
All Chlamydiae code for the core components of a T3SS. Chlamydial T3SS effector proteins should play crucial roles throughout the chlamydial developmental/infectious cycle. This includes proteins translocated across the plasma membrane - e.g., to allow entry of EBs - as well as proteins translocated across the membrane of the large vacuole (known as inclusion) that encloses EBs and/or RBs.
Chlamydial T3SS effectors are thought to modulate the host cell throughout the developmental/infectious cycle (e.g., modification of the inclusion membrane to avoid the phago-lysosomal pathway, interference with vesicular and non-vesicular trafficking for acquisition of nutrients and membrane, or inhibition of apoptosis). It is noteworthy that Chlamydia uses protein secretion mechanisms other than its T3SS to deliver effector proteins into host cells. For example, CPAF, a critical chlamydial effector protein, is not a T3S substrate; yet, how CPAF is translocated across the inclusion membrane is unknown. However, little is known about the identity and function of the vast majority of chlamydial effectors.
We are interested in identifying chlamydial T3SS effector proteins and understand their molecular and cellular mode of action.
Legionella pneumophila
Legionella pneumophila is an intracellular bacterium found ubiquitously in fresh water environments, where it parasitizes a wide range of amoebae. It is also an accidental human pathogen, as it can invade and replicate within lung macrophages giving rise to the severe type of pneumonia known as Legionnaires’ disease. Within host cells, Legionellae thrive in a remodeled compartment known as Legionella-containing vacuole (LCV) (Figure 4).
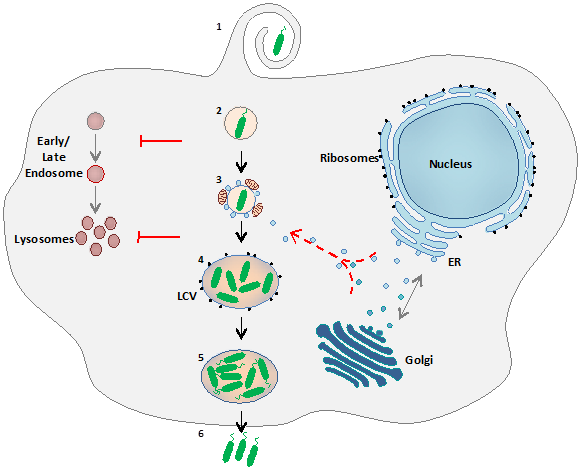
Figure 4. L. pneumophila intracellular life cycle. After uptake of the bacteria by the eukaryotic host cell (amoeba or alveolar macrophage; (1), the Legionella-containing vacuole (LCV) escapes degradation by the endocytic pathway by avoiding fusion with endosomes (2) and later delivery to lysosomes (3). The LCV interacts with mitochondria and interferes with the early secretory pathway by recruiting ER-derived vesicles trafficking to the Golgi (3), becoming a rough ER-like replicative vacuole surrounded with ribosomes (4). In this remodelled phagosome bacteria undergo several rounds of replication, become flagellated (5) and ultimately escape the host and start a new infection cycle in neighbouring cells (6).
Fundamental to the formation of a competent LCV is a Type IVB Secretion System, known as Icm/Dot (Intracellular Multiplication/Defective Organelle Trafficking), which translocates bacterial effector proteins into the host cell. A vast number of Icm/Dot substrates have been identified, and some have been linked to host cell processes such as vesicular trafficking, protein synthesis, phosphoinositide metabolism, or apoptosis. However, the function of the most effector proteins remains unknown.
We are interested in deciphering the molecular and cellular mode of action of the Legionella effector VipA, as well of other Legionella effectors that control biogenesis and trafficking of the LCV.